肝脏不同区域决定突变β-连环蛋白致癌能力
作者: aeks | 发布时间: 2025-12-08 18:04 | 更新时间: 2025-12-08 18:04
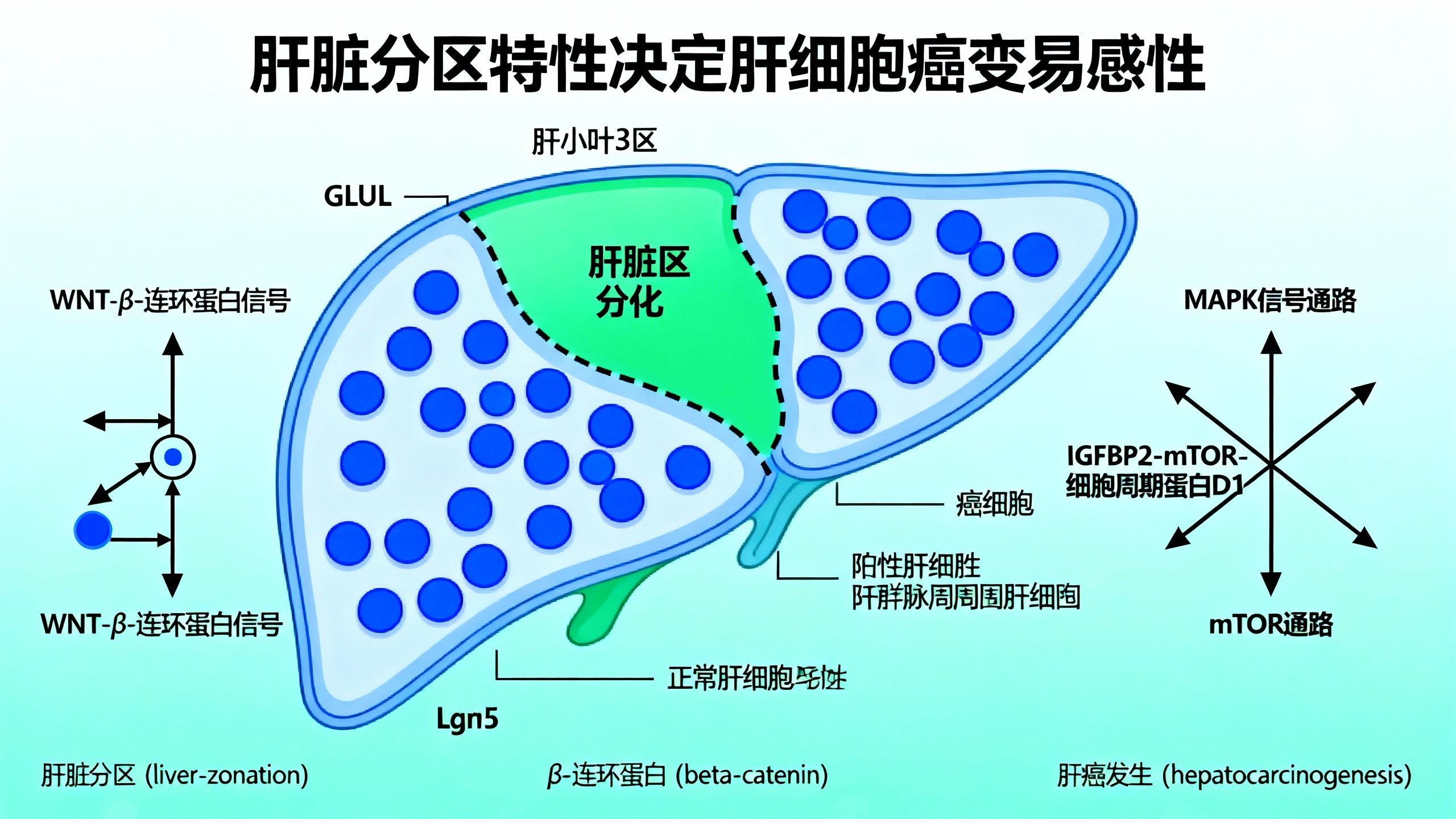
在肝脏稳态下,大多数肝细胞处于静止状态,WNT通路活性从中央静脉到门静脉呈递减梯度分布,是肝分区特性(即肝细胞根据其在肝小叶中的位置执行不同代谢功能的现象)的主要调节因子。肝小叶沿门静脉-中央静脉轴可分为三个区:1区(门管周区)、2区(小叶中区)和3区(中央静脉周区)。这种结构将核β-连环蛋白的活性限制在肝小叶内的3区肝细胞中,驱动其成熟并调控WNT靶基因的表达。相比之下,在肝脏再生和肝细胞癌(HCC)中,WNT通路活性会驱动肝细胞生长。HCC中CTNNB1基因第3外显子的点突变比其他WNT通路激活突变(如APC截断或RNF43/ZNRF3缺失)更常见,尽管后者在小鼠中已被证明具有致瘤性。了解CTNNB1突变如何促进肿瘤发生并改变分区特性非常重要,因为之前的谱系追踪研究已显示稳态肝脏中肝细胞生长以及HCC起源细胞存在分区差异。
MYC是WNT驱动的HCC所必需的。研究人员在小鼠肝脏中研究常见WNT通路突变的致瘤潜力时发现,肝细胞上皮中的散发性WNT通路突变致癌性较弱,需要很长潜伏期才能产生低肿瘤负荷。与其他常发生WNT通路突变的组织不同,通过Apc缺失激活WNT并不会增加小鼠肝脏中内源性Myc的表达。在人类HCC中,81%的CTNNB1突变肿瘤存在MYC拷贝数增加及MYC基因表达显著升高。在Ctnnb1ex3/WT肝细胞中表达R26LSL-MYC转基因可增强肿瘤发生,MYC的加入还上调了许多在Apc缺陷肠道中也常见的基因程序,这些共享基因集都与mRNA翻译和蛋白质合成相关,表明WNT驱动的癌症需要致癌性翻译组。
肿瘤发生需要适度的WNT信号。为研究散发性突变的Ctnnb1ex3/WT;R26LSL-MYC肝细胞如何发展为肿瘤,研究人员在小鼠肝脏中用低病毒滴度的AAV8.TBG.Cre处理后30和60天进行观察。分散的重组肝细胞在R26LSL-tdTomato、Ctnnb1ex3/WT和Ctnnb1ex3/WT;R26LSL-MYC肝脏中被检测到,Ctnnb1ex3/WT;R26LSL-MYC突变肝细胞还形成了由5个以上相邻细胞组成的簇(即病变)。这些病变的大小分布在30至60天之间发生变化,中等大小病变(16-25个细胞)数量增加,小病变(5-10个细胞)数量显著减少。与邻近的单个Ctnnb1ex3/WT;R26LSL-MYC突变肝细胞相比,增殖性病变的特征是WNT通路激活降低(表现为核Ctnnb1阳性率降低),但MYC表达未变。空间转录组分析显示,与单个突变肝细胞相比,增殖性病变显著富集与活性MAPK信号、mRNA翻译和蛋白质合成相关的基因集。免疫组化证实增殖病变中存在与mTOR活性和mRNA翻译增加相关的因子。IGFBP2、mTOR和CCND1活性与2区驱动的稳态增殖有关,研究发现这些因子也与WNT驱动的肿瘤发生相关,人类CTNNB1;MYC突变的HCC以及小鼠Ctnnb1ex3/WT;R26LSL-MYC肝脏肿瘤中IGFBP2显著上调。此外,病变中YAP活性增加,E-钙粘蛋白表达上调,这两者都可能解释增殖性病变中WNT通路激活降低的原因。功能验证显示,用雷帕霉素抑制mTOR活性可显著减少病变数量,短暂抑制mTOR会严重影响终末期肿瘤形成;干扰2区因子Igfbp2的表达也会减少病变;Yap和Taz缺失可阻止病变形成。这些数据表明,Ctnnb1ex3/WT;R26LSL-MYC突变肝细胞需要减弱致癌性WNT激活并激活2区特异性IGFBP2-mTOR-CCND1促生长通路才能继续肿瘤发生。
GLUL+肝细胞的致癌性较低。急性激活Ctnnb1和MYC的等位基因后,全肝细胞重组导致显著的肝细胞增殖和肝肿大。随着时间推移(诱导后4-10天),肝细胞增殖增加是短暂的,主要发生在第4天左右的GLUL-1区和2区肝细胞中,到第10天逐渐减弱;随后肝小叶轴上3区GLUL表达域显著上调,3区基因转录水平显著增加。单独过表达MYC会增加肝脏所有区域的肝细胞增殖,GLUL+和GLUL-肝细胞的增殖率相同,但本身无致瘤性。值得注意的是,R26LSL-MYC肝脏中肝细胞凋亡增加,但在Ctnnb1ex3/WT背景下凋亡受到抑制,这在人类CTNNB1;MYC突变的HCC样本中也观察到,表明WNT通路和MYC突变的协同作用在肝脏肿瘤发生中既提供增殖优势又具有抗凋亡作用。
为研究突变β-连环蛋白驱动的增殖的分区差异,对Ctnnb1ex3/WT肝脏进行全小鼠转录组空间转录组分析发现,尽管β-连环蛋白的核积累显示WNT通路激活在肝小叶中均匀分布,但GLUL+和GLUL-区域存在明显的转录分离,后者IGFBP2表达显著升高。IGFBP2是2区驱动的稳态生长所必需的,且不是直接的WNT靶基因,在Ctnnb1ex3突变肝脏中,其表达在第10天消失,与肝细胞增殖减少一致。功能实验证实,在第4天用mTOR抑制剂雷帕霉素处理或在Ifgpb2缺陷小鼠模型中诱导Ctnnb1ex3/WT;R26LSL-MYC重组,肝细胞增殖均显著减少。在3区肝细胞中过表达IGFBP2和通过AKT激活超刺激mTOR活性并未促进增殖,表明这些因子是致癌生长所必需的,但不足以诱导终末分化的3区肝细胞生长。
WNT-MYC支持增殖性翻译组。mTOR介导的mRNA翻译是肠道中WNT驱动的致癌生长的关键组成部分,2区表达的核糖体基因促进肝脏再生。对诱导后第4天(增殖期)和第10天(大部分肝细胞分化为3区命运)的肝脏进行核糖体谱分析发现,R26LSL-MYC激活增加了特定mRNA转录本的翻译效率,与Ctnnb1ex3/WT突变结合后进一步增强。在第4天高度增殖的Ctnnb1ex3/WT;R26LSL-MYC肝脏中,与细胞分裂和生长相关的转录本优先被翻译。到第10天,当肝小叶分化为以3区为主的命运时,第4天的促生长翻译组在Ctnnb1ex3/WT;R26LSL-MYC肝脏中下调,同时核糖体占据率全局降低。这些数据揭示了一个靶向RNA翻译程序,在致癌WNT和MYC激活后4天支持1区和2区肝细胞增殖所需蛋白质的产生。
Lgr5+肝细胞抵抗WNT驱动的HCC。GLUL+肝细胞对WNT驱动的增殖具有抵抗性,它们位于3区的一个静脉周围亚层。为确定3区的特定亚层是否对致癌WNT有抗性,使用一系列基因工程小鼠模型在肝小叶中进行分区限制性Cre表达。空间增殖分析显示,R26LSL-MYC重组在肝脏所有区域诱导增殖,与急性AAV8.TBG.Cre模型中MYC诱导的增殖无分区偏倚一致。在没有R26LSL-MYC转基因的情况下,Ctnnb1ex3/WT肝脏在GLUL+肝细胞附近100μm内的肝细胞增殖增加,但在中央静脉周围的GLUL+Lgr5+肝细胞中没有,这与IGFBP2表达开始的区域一致。尽管使用不同的分区Cres诱导重组,但每个模型中的增殖反应一致。为确认Lgr5特异性Ctnnb1ex3/WT和R26LSL-MYC激活没有延迟的致癌反应,在诱导后20天检查肝脏,未检测到肝肿大、异常肝细胞增殖或中央静脉处突变肝细胞的扩张。这些数据表明,极端的3区Lgr5+肝细胞命运不允许致癌WNT驱动的生长。
MAPK拮抗WNT驱动的分化。为研究分区如何影响肿瘤发生,将各种致癌突变引入Lgr5+中央静脉肝细胞并检查其致瘤潜力。中央静脉Lgr5+Ctnnb1ex3/WT;R26LSL-MYC突变体不会在肝脏中形成肿瘤,但最终会发展为肠息肉。相比之下,1区和2区Gls2Cre-ER;Ctnnb1ex3/WT;R26LSL-MYC模型确实会发展为肿瘤。先前的研究将RAS活性与门静脉(1区或2区)肝细胞联系起来,表明其可能在肝小叶1区的分区特性中起作用。引入致癌BRAF(V600E)(一种驱动RAS下游MAPK信号级联持续激活的突变蛋白激酶)可促进Lgr5+肝细胞群的肿瘤发生。由此产生的Lgr5+肝细胞来源的BrafV600E驱动的肿瘤表达1区标志物CDH1,并降低3区和WNT标志物GLUL的表达。此外,AAV8.TBG.Cre介导的全肝细胞BrafV600E突变抑制3区基因转录本并上调1区基因表达程序。将BrafV600E突变与配体依赖性Rnf43fl/fl;Znrf3fl/flWNT通路激活突变结合,显著增加器官生长并抵消分区分化,抑制3区特征的上调。以基因剂量依赖性方式增加BrafV600E会抑制WNT诱导的3区分化,并使增殖主要发生在3区GLUL+肝细胞中。降低病毒滴度以限制突变WNT通路等位基因和BrafV600E的基因重组到少数肝细胞中,会在肝脏中引发明显的肿瘤发生,并将中位生存期缩短至50-55天。由于WNT配体分泌(LGK974)和BRAF(达拉非尼)激活的组件都有小分子抑制剂,研究测试了BRAF和WNT信号在肝脏和已建立的肿瘤中是否存在动态关系。在BrafV600E/+;Rnf43fl/fl;Znrf3fl/fl癌症模型中,LGK974或达拉非尼均可抑制肿瘤发生。对达拉非尼处理的BrafV600E/+;Rnf43fl/fl;Znrf3fl/fl病变的检查显示增殖、mTOR信号和肿瘤生长减少,同时GLUL上调,CDH1和SOX9水平降低。这些结果表明,Lgr5+3区肝细胞的异常生长需要抑制3区分化的因子,WNT通路激活突变与MAPK信号结合可抑制3区分化,上调mTOR信号并显著增强肿瘤发生。
Apc缺失的致瘤性较低。突变频率分析证实CTNNB1外显子3点突变是HCC中主要的WNT通路突变。研究试图确定这是否可以用肝脏中发生的突变过程引起的散发性突变来解释,通过HCC突变特征建模预测的突变与观察到的CTNNB1热点突变比较,发现随机突变 alone 不太可能解释HCC中CTNNB1外显子3点突变的 prevalence。为检查HCC中CTNNB1突变的过度表达,在WNT-MYC肝癌模型中用Apcfl/fl等位基因替换Ctnnb1ex3/WT。在第30天检测到核Ctnnb1+Apc缺陷肝细胞,但到第60天Apcfl/fl;R26LSL-MYC肝细胞减少。与Ctnnb1ex3/WT;R26LSL-MYC模型相比,Apcfl/fl;R26LSL-MYC突变肝细胞在第30天形成更小的病变,到第60天消失。另一个关键区别是核Ctnnb1染色强度,Apcfl/fl;R26LSL-MYC突变肝细胞具有均匀、强烈的核Ctnnb1染色,而Ctnnb1ex3/WT;R26LSL-MYC肝细胞中核阳性率可变。在罕见的未分化Apcfl/fl;R26LSL-MYC终末期肿瘤中也观察到高核Ctnnb1染色,而Ctnnb1ex3/WT;R26LSL-MYC肿瘤维持较低水平的核Ctnnb1。此外,第60天Apcfl/fl;R26LSL-MYC小鼠病变形成减少,延长了生存期并降低了肿瘤发生。这些数据证实HCC对Ctnnb1ex3突变比对Apc缺失更宽容。有人提出肝细胞的高度多倍体性更倾向于单点突变而非肿瘤抑制因子缺失,Apc缺失产生的高水平致癌WNT通路激活也与肝脏中的肿瘤发生不太兼容,这与HCC中APC突变相对缺乏一致。
为进一步研究WNT通路激活水平和肝小叶分区特性如何影响肝脏肿瘤发生,使用了Apcfl/fl-hypomorphic等位基因,由于Apc水平比野生型小鼠降低30%,其基线WNT通路激活水平更高。在没有Cre的情况下,Apcfl/fl-hypomorph肝脏中WNT和3区标志物GLUL的表达增加,IGFBP2的表达最低。在这种WNT水平升高和3区扩大的背景下,在Apcfl/fl-hypomorph;R26LSL-MYC小鼠的整个肝细胞上皮中诱导基因重组,并将器官生长与另一种非hypomorphic条件性Apcfl/fl等位基因进行比较。Apcfl/fl-hypomorph;R26LSL-MYC肝脏在第4天增殖减少,导致第8天器官大小增加显著减少。在没有Cre介导的重组的情况下,Apcfl/fl-hypomorph最终会发展为肝脏肿瘤,这些在该遗传背景下产生的肿瘤WNT通路激活低,不表达核Ctnnb1和GLUL,并可变表达IGFBP2。这些发现强调,WNT高的3区肝细胞状态不允许WNT驱动的致癌生长和肝脏中的肿瘤发生。