无需实验试错,AI直接设计能结合药物的蛋白质
作者: aeks | 发布时间: 2026-06-25 18:02 | 更新时间: 2026-06-25 18:02
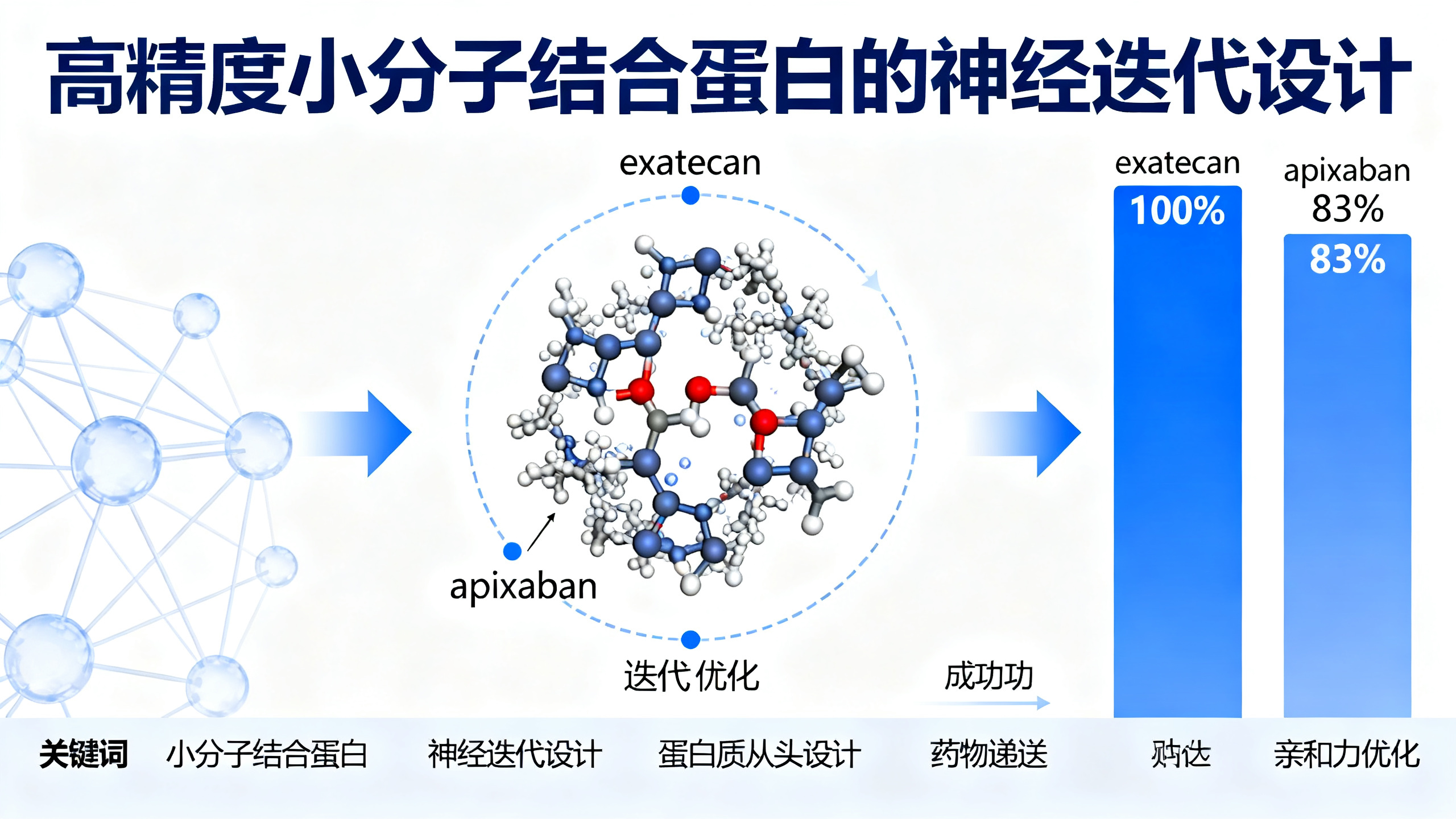
无需实验试错,AI直接设计能结合药物的蛋白质
本文介绍了一种名为‘神经迭代筛选–扩展’(NISE)的全新计算方法,用于从头设计能特异性结合小分子药物的蛋白质。传统方法面临巨大挑战:需同时优化蛋白质序列、三维结构和小分子配体构象,而现有深度学习算法难以兼顾这三者。NISE巧妙地将两个神经网络(LASErMPNN序列设计网络和RFAA/Boltz-2蛋白–配体复合物结构预测网络)组成闭环系统,反复进行‘设计—预测—筛选—再设计’的迭代:先用LASErMPNN为给定蛋白骨架和配体位置设计兼容的氨基酸序列;再用结构预测网络评估该序列能否形成稳定、精准的蛋白–配体复合物;然后挑选预测置信度最高的几个结果,作为下一轮设计的起点。这种双向优化显著优于依赖物理能量函数的传统方法。研究团队用NISE成功设计出两类全新蛋白:一类专一结合抗癌药exatecan(成功率100%,最强结合体亲和力达120纳摩尔),另一类结合抗凝药apixaban(成功率83%,最强结合体亲和力达80皮摩尔)。其中exatecan结合蛋白(EPIC)不仅能高精度识别药物,还能像‘保护罩’一样包裹住药物中易水解的活性基团(内酯环),使其在生理环境中稳定数天——这是现有技术无法实现的关键功能。更令人惊喜的是,仅靠LASErMPNN神经网络‘校对’原始序列,就无需任何实验反馈,仅通过两次氨基酸替换,就将EPIC对exatecan的亲和力提升了100倍。整个过程逻辑清晰、步骤明确,展示了人工智能如何真正赋能精准药物蛋白设计,为未来药物递送、生物传感和人工催化等领域提供了强大通用工具。