新疗法降低胆固醇近一半,无需他汀药也无副作用
作者: aeks | 发布时间: 2025-10-23 13:59 | 更新时间: 2025-10-23 13:59
学科分类: 临床医学 基础医学 生物化学与分子生物学 药学
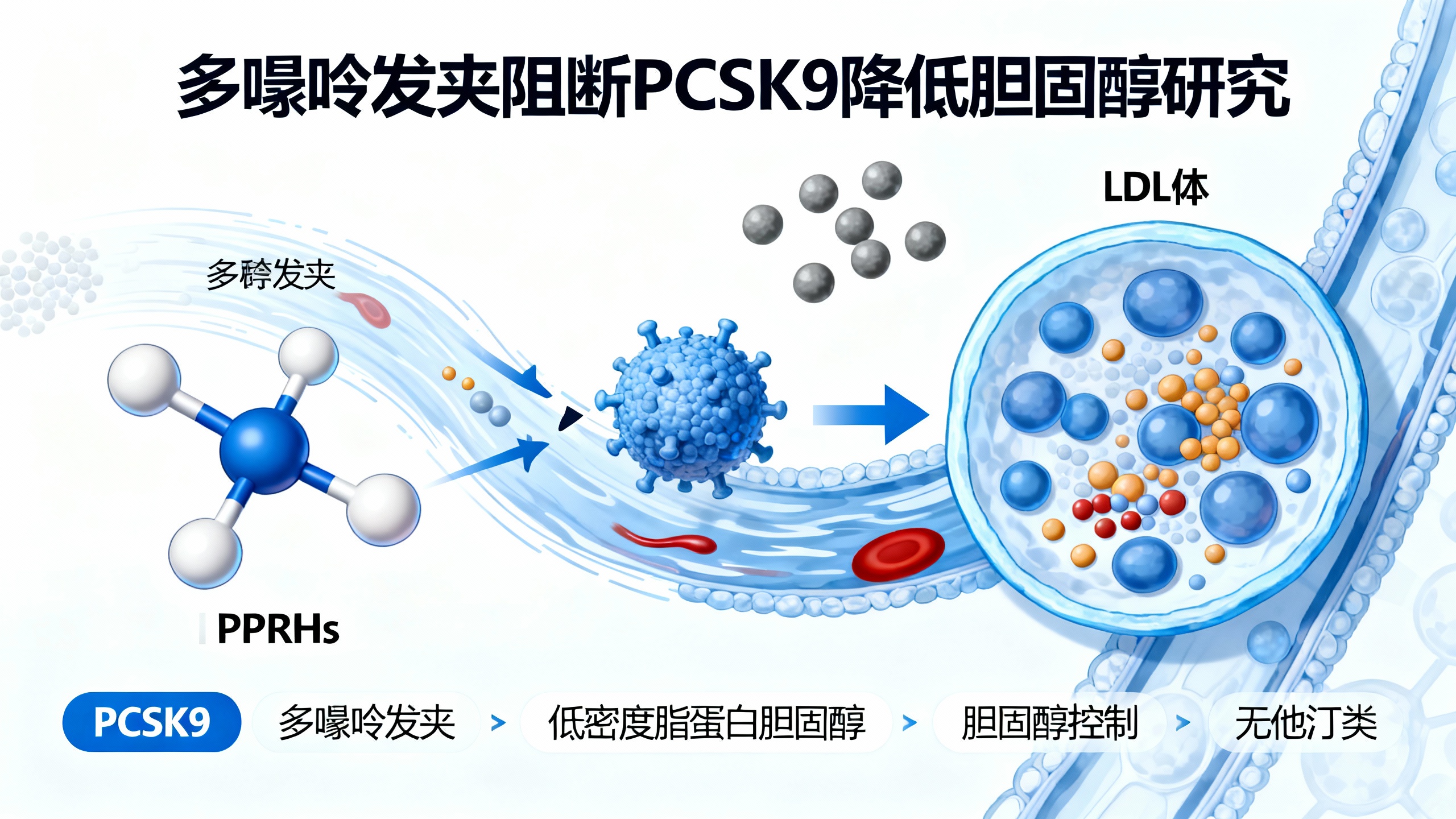
研究团队研发出一种阻断PCSK9活性的新策略。PCSK9是一种在控制血液中低密度脂蛋白胆固醇(LDL-C,常称“坏”胆固醇)含量方面起关键作用的蛋白质。这种创新方法依赖于名为多嘌呤发夹(PPRHs)的分子,能帮助细胞吸收更多胆固醇,防止其在动脉中堆积,且不会产生他汀类药物常见的副作用。
该研究发表在《Biochemical Pharmacology》杂志,由巴塞罗那大学药学院与食品科学学院及纳米科学与纳米技术研究所(IN2UB)的Carles J. Ciudad教授和Verònica Noé教授领衔,与美国波特兰俄勒冈大学的Nathalie Pamir合作完成,资金来源于西班牙科学、创新与大学部(MICINN)和美国国立卫生研究院(NIH)。
靶向PCSK9蛋白:过去十年,PCSK9(前蛋白转化酶枯草溶菌素/kexin 9型)已成为胆固醇治疗和心血管保护的主要靶点。这种酶会与细胞表面通常捕获LDL胆固醇的受体结合,减少受体数量,导致血液中LDL胆固醇水平升高,增加高胆固醇血症风险。
团队开发的新技术利用PPRHs阻止特定基因转录,有效沉默其表达。本研究中,PPRHs被用于抑制PCSK9基因,从而增加低密度脂蛋白受体(LDLR),改善细胞对胆固醇的摄取,有助于降低循环胆固醇水平和动脉斑块堆积风险。
多嘌呤发夹的作用机制:PPRHs是单链DNA寡核苷酸,能精确结合互补的DNA或RNA序列。研究首次证实,HpE9和HpE12这两种特定PPRHs可降低PCSK9的RNA和蛋白水平,同时提高LDLR水平。生物化学与生理学系的Carles J. Ciudad教授指出,HpE9和HpE12的每条链中的一个臂,分别通过沃森-克里克键与PCSK9外显子9和12的多嘧啶序列特异性结合,这种结合会抑制基因转录、RNA聚合酶作用或转录因子结合。
该新疗法已在表达人类PCSK9基因的转基因小鼠体内得到验证。Verònica Noé教授表示,HpE9和HpE12在HepG2细胞中均高度有效,其中HpE12使PCSK9的RNA水平降低74%、蛋白水平降低87%;在转基因小鼠中,单次注射HpE12后第三天,血浆PCSK9水平降低50%,胆固醇水平降低47%。
迈向无他汀类胆固醇控制:自PCSK9被确定为降低血浆胆固醇治疗的重要靶点后,已出现多种降低或阻断其作用的疗法,如siRNA、反义寡核苷酸或CRISPR基因沉默技术,其中针对PCSK9的siRNA药物Inclisiran及单克隆抗体(如依洛尤单抗、阿利西尤单抗)较为突出。专家们总结,PPRHs(尤其是HpE12)是具有诸多优势的治疗性寡核苷酸,合成成本低、稳定性好且无免疫原性,基于PPRHs的PCSK9阻断方法不会产生他汀类药物相关的肌病等副作用。