用深度学习实现核磁共振的“超清”成像
作者: aeks | 发布时间: 2026-04-22 12:02 | 更新时间: 2026-04-22 12:02
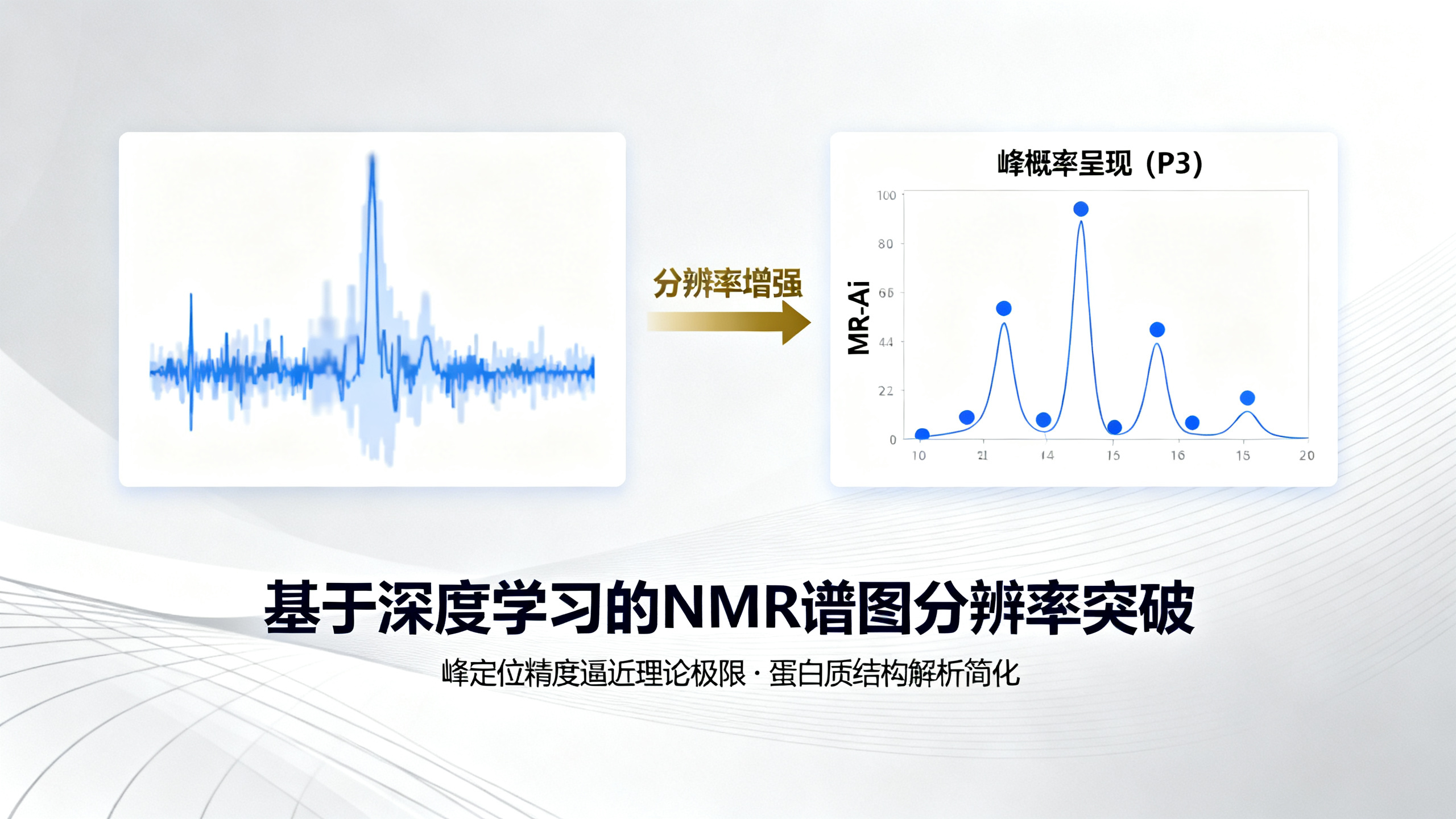
本文介绍了一种突破性的核磁共振(NMR)谱图分析新方法,旨在解决生物大分子NMR研究中长期存在的核心难题——谱图分辨率不足。传统NMR谱图以信号强度展示(即“传统强度呈现”,TIP),但易受噪声、信号重叠、动态范围过大及伪峰干扰,尤其在分析大型或无序蛋白(如Tau、MALT1)时,峰难以区分,严重阻碍蛋白质结构与动力学研究。为此,作者团队创新性地提出“峰概率呈现”(P3):它不直接显示强度,而是为谱图中每一个数据点赋予一个0–1之间的概率值,表示该点是真实峰顶的可能性。这一概念将分辨率问题转化为统计分类任务——判断“此处是否有峰”。实现P3的核心是一个名为MR-Ai的新型深度学习模型。该模型设计精巧:它不处理整张高维谱图,而是聚焦于每个待测点周围的“十字形”局部区域(模拟NMR信号的物理本质),通过轻量级网络(仅约8000个参数)高效提取关键特征(如峰形、相位畸变、噪声模式等),最终输出高置信度的概率预测。P3的优势体现在三方面:第一,超高分辨率——实验表明,P3可将峰宽压缩至1–2个数据点,远超传统傅里叶变换谱图的理论极限(2–3个点),在MALT1和Tau等复杂蛋白谱图中成功分离出原本完全重叠的峰;第二,超低动态范围——传统谱图中强峰可能比弱峰亮数十倍,导致弱峰被掩盖,而P3中不同强度的峰概率值接近(如均在0.8左右),使所有可靠信号一目了然,极大简化人工判读;第三,智能抗噪——P3能有效抑制t1噪声、截断伪峰及非均匀采样(NUS)残留伪影等常见干扰。研究团队用60种不同大小与复杂度的蛋白质谱图(来自100-蛋白数据库)进行了大规模验证,结果证明P3方法稳定可靠,即使在仅5%–10%数据量的极端稀疏采样下,其峰识别准确率(F1分)仍远高于现有主流工具ARTINA。为进一步定量验证,作者还构建了大量合成谱图(含精确已知的峰位置与噪声),证实P3的峰定位精度已非常接近由信息论决定的“克拉美-罗下界”(CRLB)这一理论天花板。此外,该方法还支持“多谱图协同处理”,例如利用高灵敏度的2D谱作为“支持谱”来辅助解析质量较差的3D谱,这在实时实验监控(靶向采集,TA)中尤为实用,可大幅缩短昂贵的NMR实验时间。总之,P3并非对传统方法的小修小补,而是借助深度学习重构了NMR谱图的表达范式,将物理模型、统计推断与人工智能深度融合,为获取蛋白质原子级细节提供了更清晰、更鲁棒、更智能的新工具。