激活的ATF6α蛋白:肝脏肿瘤的“推手”,削弱免疫系统监视能力
作者: aeks | 发布时间: 2026-02-06 06:04 | 更新时间: 2026-02-06 06:04
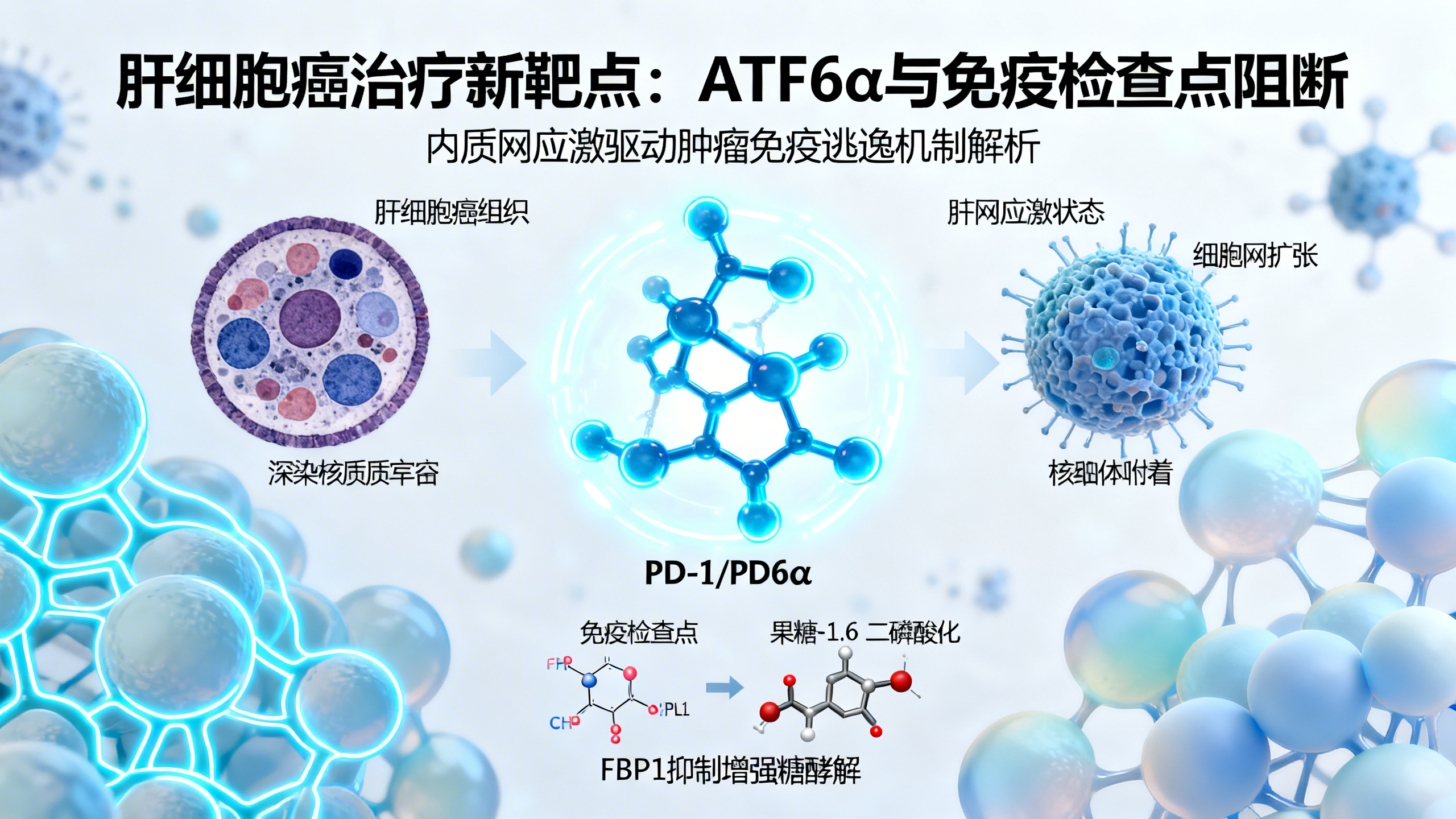
肝细胞癌(HCC)占原发性肝癌的80%-85%,主要由慢性肝炎中发生恶性转化的肝细胞发展而来。尽管免疫疗法改善了患者生存,但复杂的遗传、代谢和炎症相互作用仍是有效治疗的障碍。内质网(ER)应激和未折叠蛋白反应(UPR)是癌症的不良预后因素,其中UPR包含PERK、IRE1α和ATF6α三种跨膜蛋白。以往认为ATF6α在急性ER应激中起适应性作用,而本研究发现其持续激活会成为肝癌的驱动因子和代谢主调节因子,促进免疫抑制。
在人类HCC中,ATF6α激活与侵袭性肿瘤表型显著相关,表现为患者生存率降低、肿瘤进展加快和局部免疫抑制。通过对22个HCC数据集分析,ATF6α的mRNA及其激活特征在肿瘤组织中显著升高,且与患者生存缩短相关。免疫组化显示,高ATF6α激活的HCC样本多为高级别(G3)肿瘤,且肿瘤中活化的核ATF6α(nATF6α)积累增加。空间转录组学和成像质谱流式细胞术(IMC)发现,ATF6α高表达区域富集PD-1/PD-L1信号、缺氧、细胞周期和糖酵解相关基因,同时糖异生关键酶FBP1表达降低,CD8+T细胞浸润增加但处于耗竭状态。
在小鼠模型中,肝细胞特异性ATF6α激活诱导进行性肝炎,伴随ER应激、免疫抑制和肝细胞增殖。机制上,活化的ATF6α直接结合FBP1基因调控元件并抑制其表达,导致糖酵解增强、糖原和葡萄糖耗竭。恢复FBP1表达可减轻ATF6α激活相关病理。长期ATF6α激活引发肝癌,肿瘤内T细胞浸润但因营养剥夺而耗竭。免疫检查点阻断(ICB)可恢复免疫监视并减少HCC,且对免疫治疗完全应答的HCC患者ATF6α激活水平显著更高。
通过基因敲除、肝细胞特异性敲除或反义寡核苷酸(ASO)靶向Atf6,在临床前肝癌模型中均能抑制HCC。综上,ATF6α持续激活驱动ER应激,通过糖酵解依赖性免疫抑制促进肝癌,并使肿瘤对ICB敏感。该研究表明,持续激活的ATF6α是肝癌驱动因子、ICB应答的潜在分层标志物和治疗靶点。
标签: 免疫检查点阻断 内质网应激 果糖-1,6-二磷酸酶 肝细胞癌